武汉八颗星生物科技有限公司拥有成熟的传统一代Sanger BAC末端测序技术及平台,BAC末端序列(BES)长度平均达到700 bp。
大片段DNA 文库如Fosmid和BAC文库常常被应用于鉴定结构变异或组装错误、填补基因组scaffold内的空隙以及辅助全基因组组装。这些文库的成对末端序列具有较大的跨度,可以有效解决复杂物种基因组中长的重复序列的定位问题,及其在全基因组组装中提供大范围的关联信息。
目前,已有一些利用大片段DNA文库的高通量末端测序方法发表,例如Fosill和pBACode。另一些不依赖于大片段DNA文库的高通量末端测序方法也发展迅速,例如10X Genomics、Hi-C和BioNano等。但这些方法均是基于NGS测序平台的,得到的末端读长无法超过1 kb。
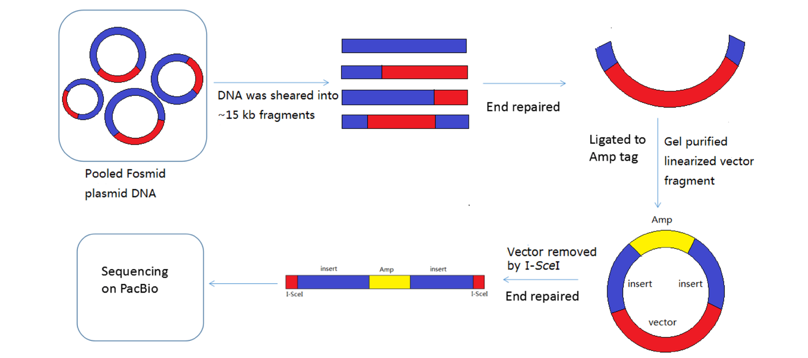
我们开发了一种基于PacBio平台的BAC末端测序方法。该方法将大片段DNA文库的长跨度与三代测序技术的长读长优势结合起来,产生成对的末端,平均每端的长度达2-3 kb,远远超过目前所有的末端测序技术得到的末端长度。不仅如此,通过一系列技术优化后,成对长末端正确率高达95.3% (请见详细信息)。
该方法简单易行,可与其他方法互补,共同用于组装复杂的基因组,检测结构变异和装配错误以及评估装配质量。
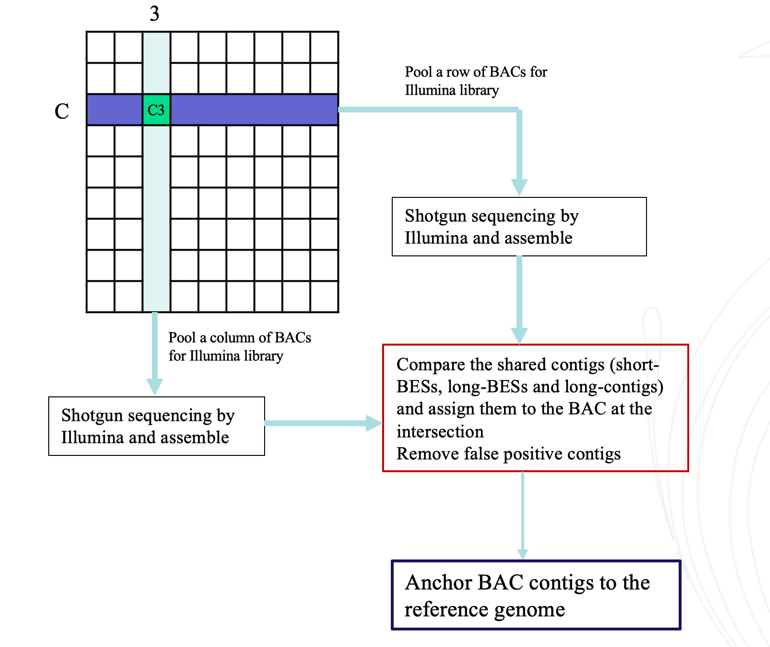
克隆矩阵混合池鸟枪法最早由Cai等提出并应用到恒河猴基因组测序中(Cai等. 2001),其主要思想是:BAC克隆首先被排列成二维矩阵的形式,分别将每一行和每一列所有克隆混合在一起,构建亚克隆文库并测序。通过比对行池和列池亚克隆文库的测序序列将序列分配到交叉位点上,然后组装出每个交叉位点的BAC序列,再通过人工校正形成参考基因组序列。
我们结合克隆矩阵混合池测序策略和Illumina测序技术,从BAC混合池测序序列中解析出BAC末端序列,通过解析的末端序列,将BAC克隆定位到参考基因组上。(请见详细信息)。
以往,为了将BAC克隆定位到基因组上,常利用Sanger测序法对BAC克隆进行逐个双末端测序,这种方法适用于少量BAC的定位。我们的方法可同时将近万个BAC克隆定位到基因组上,成功率高达89.65%,这极大的降低了成本,缩短了实验周期。
BAC文库与基因组的整合有利于充分发挥文库的价值,为后续的基因组序列校正和完善提供可靠保障,同时为图位克隆、分子育种和基因组功能研究等奠定基础。